Generate single-cell images¶
Here, we are going to process the previously ingested microscopy images with the scPortrait pipeline to generate single-cell images that we can use to assess autophagosome formation at a single-cell level.
import lamindb as ln
from collections.abc import Iterable
from pathlib import Path
from scportrait.pipeline.extraction import HDF5CellExtraction
from scportrait.pipeline.project import Project
from scportrait.pipeline.segmentation.workflows import CytosolSegmentationCellpose
ln.track()
Query microscopy images¶
First, we query for the raw and annotated microscopy images.
input_images = ln.Artifact.filter(
ulabels__name="autophagy imaging", description__icontains="raw image", suffix=".tif"
)
The experiment includes two genotypes (WT and EI24KO) under two treatment conditions (unstimulated vs. 14h Torin-1).
Multiple clonal cell lines were imaged for each condition across several fields of view (FOVs) and imaging channels.
We’ll extract single-cell images from each FOV and annotate them with metadata including genotype, treatment condition, clonal cell line, and imaging experiment.
input_images_df = input_images.df(features=True)
display(input_images_df.head())
conditions = input_images_df["stimulation"].unique().tolist()
cell_line_clones = input_images_df["cell_line_clone"].unique().tolist()
FOVs = input_images_df["FOV"].unique().tolist()
Alternatively, we can query for the ULabel directly:
conditions = ln.ULabel.filter(
links_artifact__feature__name="stimulation", artifacts__in=input_images
).distinct()
cell_line_clones = ln.ULabel.filter(
links_artifact__feature__name="cell_line_clone", artifacts__in=input_images
).distinct()
FOVs = ln.ULabel.filter(
links_artifact__feature__name="FOV", artifacts__in=input_images
).distinct()
By iterating through conditions, cell lines and FOVs, we should only have 3 images showing a single FOV to enable processing using scPortrait.
# Create artifact type feature and associated label
ln.Feature(name="artifact type", dtype=ln.ULabel).save()
ln.ULabel(name="scportrait config").save()
# Load config file for processing all datasets
config_file_af = ln.Artifact.using("scportrait/examples").get(
key="input_data_imaging_usecase/config.yml"
)
config_file_af.description = (
"config for scportrait for processing of cells stained for autophagy markers"
)
config_file_af.save()
# Annotate the config file with the metadata relevant to the study
config_file_af.features.add_values(
{"study": "autophagy imaging", "artifact type": "scportrait config"}
)
Process images with scPortrait¶
Let’s take a look at the processing of one example FOV.
# Get input images for one example FOV
condition, cellline, FOV = conditions[0], cell_line_clones[0], FOVs[0]
images = (
input_images.filter(ulabels=condition)
.filter(ulabels=cellline)
.filter(ulabels=FOV)
.distinct()
)
# Quick sanity check - all images should share metadata except channel/structure
values_to_ignore = ["channel", "imaged structure"]
features = images.first().features.get_values()
shared_features = {k: v for k, v in features.items() if k not in values_to_ignore}
for image in images:
image_features = image.features.get_values()
filtered_features = {
k: v for k, v in image_features.items() if k not in values_to_ignore
}
assert shared_features == filtered_features
# Get image paths in correct channel order
input_image_paths = [
images.filter(ulabels__name=channel).one().cache()
for channel in ["DAPI", "Alexa488", "mCherry"]
]
# Create output directory and unique project ID
output_directory = "processed_data"
unique_project_id = f"{shared_features['cell_line_clone']}/{shared_features['stimulation']}/{shared_features['FOV']}".replace(
" ", "_"
)
project_location = f"{output_directory}/{unique_project_id}/scportrait_project"
# Create directories
Path(project_location).mkdir(parents=True, exist_ok=True)
# Initialize the scPortrait project
project = Project(
project_location=project_location,
config_path=config_file_af.cache(),
segmentation_f=CytosolSegmentationCellpose,
extraction_f=HDF5CellExtraction,
overwrite=True,
)
# Load images and process
project.load_input_from_tif_files(
input_image_paths, overwrite=True, channel_names=["DAPI", "Alexa488", "mCherry"]
)
project.segment()
project.extract()
Let’s look at the input images we processed.
project.plot_input_image()
WARNING:ome_zarr.io:version mismatch: detected: RasterFormatV02, requested: FormatV04
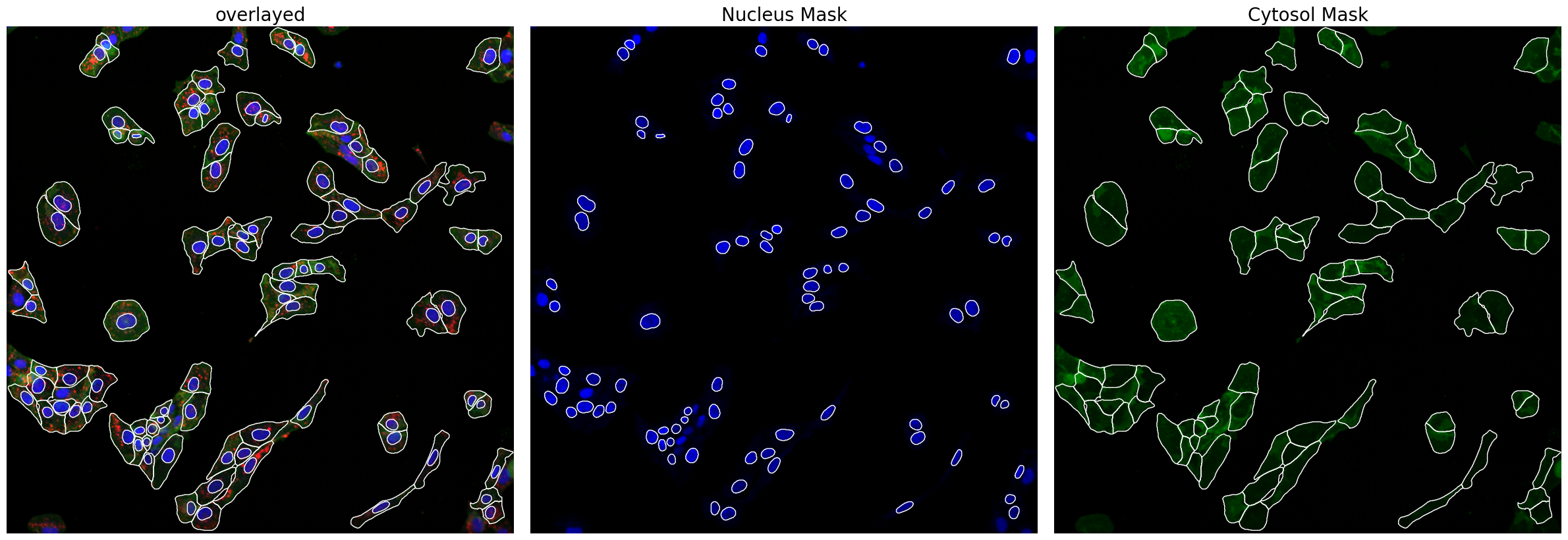
Now we can look at the results generated by scPortrait. First, the segmentation masks.
project.plot_segmentation_masks()
WARNING:ome_zarr.io:version mismatch: detected: RasterFormatV02, requested: FormatV04
And then extraction results consisting of individual single-cell images over all of the channels.
project.plot_single_cell_images()

Save and annotate results¶
Now we also want to save these results to the instance.
ln.Artifact.from_spatialdata(
sdata=project.filehandler.get_sdata(),
description="scportrait spatialdata object containing results of cells stained for autophagy markers",
key=f"processed_data_imaging_use_case/{unique_project_id}/spatialdata.zarr",
).save()
# Define schemas for single-cell image dataset
schemas = {
"var.T": ln.Schema(
name="single-cell image dataset schema var",
description="column schema for data measured in obsm[single_cell_images]",
itype=ln.Feature,
).save(),
"obs": ln.Schema(
name="single-cell image dataset schema obs",
features=[
ln.Feature(name="scportrait_cell_id", dtype="int", coerce_dtype=True).save()
],
).save(),
"uns": ln.Schema(
name="single-cell image dataset schema uns",
itype=ln.Feature,
dtype=dict,
).save(),
}
# Create composite schema
h5sc_schema = ln.Schema(
name="single-cell image dataset",
otype="AnnData",
slots=schemas,
).save()
# Curate the AnnData object
curator = ln.curators.AnnDataCurator(project.h5sc, h5sc_schema)
curator.validate()
# Save artifact with annotations
artifact = curator.save_artifact(
key=f"processed_data_imaging_use_case/{unique_project_id}/single_cell_data.h5ad"
)
# Add metadata and labels
annotation = shared_features.copy()
annotation["imaged structure"] = [
ln.ULabel.using("scportrait/examples").get(name=name)
for name in ["LckLip-mNeon", "DNA", "mCherry-LC3B"]
]
artifact.features.add_values(annotation)
artifact.labels.add(ln.ULabel(name="scportrait single-cell images").save())
! 5 terms not validated in feature 'columns' in slot 'var.T': '0', '1', '2', '3', '4'
→ fix typos, remove non-existent values, or save terms via: curator.slots['var.T'].cat.add_new_from('columns')
! no values were validated for columns!
! 1 term not validated in feature 'keys' in slot 'uns': 'single_cell_images'
→ fix typos, remove non-existent values, or save terms via: curator.slots['uns'].cat.add_new_from('keys')
! no values were validated for keys!
→ returning existing schema with same hash: Schema(uid='0000000000000000', name='single-cell image dataset schema var', description='column schema for data measured in obsm[single_cell_images]', n=-1, is_type=False, itype='Feature', hash='kMi7B_N88uu-YnbTLDU-DA', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, space_id=1, created_by_id=1, run_id=3, created_at=2025-07-14 06:41:47 UTC)
→ returning existing schema with same hash: Schema(uid='fxdP2u6cVNyhRg6Y', name='single-cell image dataset schema obs', n=1, is_type=False, itype='Feature', hash='98jtXAU6e8cL9NqcucBT8w', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, space_id=1, created_by_id=1, run_id=3, created_at=2025-07-14 06:41:48 UTC)
→ returning existing schema with same hash: Schema(uid='0000000000000000', name='single-cell image dataset schema var', description='column schema for data measured in obsm[single_cell_images]', n=-1, is_type=False, itype='Feature', hash='kMi7B_N88uu-YnbTLDU-DA', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, space_id=1, created_by_id=1, run_id=3, created_at=2025-07-14 06:41:47 UTC)
To process all files in our dataset efficiently, we’ll create a custom image processing function.
We decorate this function with :func:~lamindb.tracked to track data lineage of the input and output artifacts.
The function will skip files that have already been processed and uploaded, improving processing time by avoiding redundant computations.
@ln.tracked()
def process_images(
config_file_af: ln.Artifact,
input_artifacts: Iterable[ln.Artifact],
h5sc_schema: ln.Schema,
output_directory: str,
) -> None:
# Quick sanity check - all images should share metadata except channel/structure
values_to_ignore = ["channel", "imaged structure"]
first_features = input_artifacts.first().features.get_values()
shared_features = {
k: v for k, v in first_features.items() if k not in values_to_ignore
}
for artifact in input_artifacts:
artifact_features = artifact.features.get_values()
filtered_features = {
k: v for k, v in artifact_features.items() if k not in values_to_ignore
}
assert shared_features == filtered_features
# Create a unique project ID
unique_project_id = f"{shared_features['cell_line_clone']}/{shared_features['stimulation']}/{shared_features['FOV']}".replace(
" ", "_"
)
# Check if already processed
base_key = f"processed_data_imaging_use_case/{unique_project_id}"
try:
ln.Artifact.using("scportrait/examples").get(
key=f"{base_key}/single_cell_data.h5ad"
)
ln.Artifact.using("scportrait/examples").get(key=f"{base_key}/spatialdata.zarr")
print("Dataset already processed. Skipping.")
return
except ln.Artifact.DoesNotExist:
pass
# Get image paths in channel order
input_image_paths = [
input_artifacts.filter(ulabels__name=channel).one().cache()
for channel in ["DAPI", "Alexa488", "mCherry"]
]
# Setup and process project
project_location = f"{output_directory}/{unique_project_id}/scportrait_project"
Path(project_location).mkdir(parents=True, exist_ok=True)
project = Project(
project_location=project_location,
config_path=config_file_af.cache(),
segmentation_f=CytosolSegmentationCellpose,
extraction_f=HDF5CellExtraction,
overwrite=True,
)
project.load_input_from_tif_files(
input_image_paths, overwrite=True, channel_names=["DAPI", "Alexa488", "mCherry"]
)
project.segment()
project.extract()
# Save single-cell images
curator = ln.curators.AnnDataCurator(project.h5sc, h5sc_schema)
artifact = curator.save_artifact(key=f"{base_key}/single_cell_data.h5ad")
annotation = shared_features.copy()
annotation["imaged structure"] = [
ln.ULabel.using("scportrait/examples").get(name=name)
for name in ["LckLip-mNeon", "DNA", "mCherry-LC3B"]
]
artifact.features.add_values(annotation)
artifact.labels.add(ln.ULabel.get(name="scportrait single-cell images"))
# Save SpatialData object
ln.Artifact.from_spatialdata(
sdata=project.filehandler.get_sdata(),
description="scportrait spatialdata object containing results of cells stained for autophagy markers",
key=f"{base_key}/spatialdata.zarr",
).save()
ln.Param(name="output_directory", dtype="str").save()
Now we are ready to process all of our input images and upload the generated single-cell image datasets back to our instance.
for condition in conditions:
for cellline in cell_line_clones:
for FOV in FOVs:
images = (
input_images.filter(ulabels=condition)
.filter(ulabels=cellline)
.filter(ulabels=FOV)
.distinct()
)
if images:
process_images(
config_file_af,
input_artifacts=images,
h5sc_schema=h5sc_schema,
output_directory=output_directory,
)
example_artifact = ln.Artifact.filter(
ulabels=ln.ULabel.get(name="scportrait single-cell images")
).first()
example_artifact.view_lineage()

ln.finish()